一种催化裂化柴油加氢生产清洁汽油的方法与流程-j9九游会真人
1.本发明涉及石油化工技术领域,具体涉及一种催化裂化柴油加氢生产清洁汽油的方法。
背景技术:
2.柴油中的芳烃以重质芳烃为主,这类重质芳烃燃烧性能差,是柴油车尾气pm2.5颗粒物排放的主要原因,尤其是多环芳烃,具有致癌性、致诱变性,是主要的污染物之一。在发展低碳经济、循环经济、实现可持续发展的形势下,油品质量升级步伐明显加快,对柴油中芳烃含量的要求也越来越严格。生产清洁油品成为目前炼油工业发展的重点之一。原油以重质、劣质原油为主,二次加工以催化裂化、焦化为主,生产的催化裂化、焦化柴油芳烃含量高,成为制约相关单位生产高标准清洁汽油的瓶颈,如何降低柴油中芳烃含量成为困扰炼厂的一个难题。
3.为保护环境,世界各国对发动机燃料的组成提出了更严格的限制,以降低有害物质的排放。其中对汽油中的硫和烯烃含量的限制更为苛刻。汽油质量的主要问题是硫含量和烯烃含量高。这主要有两方面的原因:一是催化裂化的加工能力占二次加工的比例较大,催化裂化汽油是商品汽油的主要来源,其他汽油组分少,调和能力差。从汽油池中各种调和组分看,催化裂化汽油中的硫和烯烃是汽油池中硫和烯烃主要来源;特别是沿江地区炼油厂主要加工硫含量高的胜利原油,沿海地区炼油厂加工进口高硫原油量也在不断上升,导致汽油池中硫含量不断上升。二是催化裂化加工的原料向重质化方向发展,随着渣油掺入量的进一步增加,催化裂化汽油中硫含量、烯烃含量将继续维持在较高的水平。因此降低催化裂化汽油中硫和烯烃含量将成为车用汽油清洁化的主要途径。
4.降低催化裂化汽油的硫含量通常可采用催化裂化原料加氢处理或者催化裂化汽油加氢脱硫两种技术方案。催化裂化原料加氢处理装置需要在温度和压力都很苛刻的条件下操作,而且处理量大,氢耗大,这将提高装置的投资,而该技术方案对降低催化裂化汽油的烯烃含量作用有限。传统的催化裂化汽油加氢脱硫技术虽然可以大幅度地降低硫含量和烯烃含量,但由于加氢过程中将具有高辛烷值的烯烃组分大量饱和,致使汽油产品辛烷值损失很大。
5.us5411658公开了一种汽油改质方法,该方法先将fcc汽油加氢精制后,直接进入第二段,进行辛烷值恢复过程的一种处理fcc汽油的方法。该专利加氢精制采用传统加氢精制催化剂,辛烷值恢复过程采用β-沸石催化剂。但该方法所使用原料油终馏点偏高。
6.]us6334948公开了一种生产低硫汽油的方法,该方法先将全馏分催化裂化汽油切割成轻重两种馏分,轻馏分使用镍基催化剂加氢脱硫;重馏分进行选择性加氢脱硫,然后将精制后的轻重馏分混合得到全馏分产品。
7.us6495030公开了一种催化裂化汽油脱硫的工艺方法。该方法至少有两个催化蒸馏反应器,第一个催化蒸馏反应器在精馏段装填硫醚化催化剂,第二个催化蒸馏反应器在精馏段装填加氢脱硫催化剂。催化裂化汽油原料先在第一催化蒸馏反应器内分馏成轻馏分
和较重的馏分,当轻馏分通过硫醚化催化剂时,将所含的硫醇硫转化为重的硫化物,并与较重馏分一起从塔底流出进入第二催化蒸馏反应器。物流在第二催化蒸馏反应器中分馏成中间馏分和重馏分,中间馏分通过精馏段进行加氢脱硫,重馏分从塔底流出进入第三催化蒸馏塔或加氢脱硫反应器中进行脱硫,最后将处理后的轻、中、重馏分混合得到全馏分产品。该方法仅提供了一种降低催化裂化汽油中硫含量的方法,没有考虑加氢过程烯烃饱和后,辛烷值损失的问题。而且流程复杂,操作不便。
技术实现要素:
8.本发明的目的在于提出一种催化裂化柴油加氢生产清洁汽油的方法,采用加氢精制、异构化、裂解反应,以催化裂化柴油为原料,生产高芳烃含量的优质清洁汽油,促进催化裂化柴油完全转化,产品质量优良,产品收率高,汽油馏分具有低的硫含量和烯烃含量,其中芳烃含量高,主要为单环芳烃,脱除了大部分多环芳烃,作为高辛烷值汽油产品,同时制备过程中,整体能耗低,降低了设备投资,显著提升催化裂化柴油价值,具有广泛的应用前景。
9.本发明的技术方案是这样实现的:本发明提供一种催化裂化柴油加氢生产清洁汽油的方法,包括以下步骤:(1)将催化裂化柴油与氢气混合,在加氢精制催化剂的催化作用下,进行加氢精制反应;(2)将步骤(1)获得的生成油与氢气混合,在加氢异构化催化剂的催化作用下,进行加氢异构化反应;(3)将步骤(2)获得的生成油在催化裂解催化剂作用下发生催化裂解反应,生成低碳烯烃和汽油馏分,产物中》200℃的馏分重新返回步骤(2)进行催化转化;(4)收集步骤(3)中的汽油馏分混合均匀后,得到合格清洁汽油产品;所述催化裂化柴油原料的沸点范围为165-400℃,总芳烃含量高于60wt%;所述加氢精制催化剂为b元素的掺杂的mo-w沉积褶皱石墨包覆纳米球催化剂;所述加氢异构化催化剂为ni-v的磷化物沉积改性hy型分子筛催化剂;所述催化裂解催化剂为pt-ni金属沉积改性hy型分子筛催化剂活性组分与氧化铝基质材料的混合物制得的催化剂。
10.作为本发明的进一步改进,所述加氢精制催化剂的制备方法如下:s1.sio2纳米球的制备:将正硅酸烷基酯溶于有机溶剂中,加入含有表面活性剂的水溶液中,调节溶液ph值,乳化,加热搅拌反应,离心,洗涤,干燥,制得sio2纳米球;优选地,所述有机溶剂选自石油醚、乙酸乙酯、乙酸甲酯、乙酸丁酯、甲苯、二甲苯、环己烷、正己烷中的至少一种。所述表面活性剂选自十二烷基苯磺酸钠、十二烷基硫酸钠、十二烷基磺酸钠、十四烷基苯磺酸钠、十四烷基磺酸钠、十四烷基硫酸钠、十六烷基磺酸钠、十六烷基苯磺酸钠、十八烷基苯磺酸钠、十八烷基磺酸钠中的至少一种;s2.褶皱氧化石墨包覆纳米球的制备:将氧化石墨溶于水中,加入步骤s1制得的sio2纳米球,搅拌混合均匀,喷雾干燥,制得褶皱氧化石墨包覆纳米球;s3.mo-w金属的沉积:将浓硝酸加入硝酸钼溶液中,得到a液;将钨酸铵溶于水中,得到b液;将a液加入b液中,加入步骤s2制得的褶皱氧化石墨包覆纳米球,搅拌进行共沉淀
80℃,时间为2-4h,所述焙烧的温度为500-600℃,时间为1-3h;步骤u2中所述硝酸镍、氯化铂、改性hy型分子筛的质量比为5-7:3-5:25-30,所述氢气还原以5-7℃/min的升温速率升温至400-500℃下恒温还原2-4h;步骤u3中所述焙烧的温度为450-550℃,时间为1-3h,所述基质材料和催化剂活性成分的质量比为50-60:5-7。
17.作为本发明的进一步改进,所述加氢精制反应的氢分压5-12mpa,反应温度240-400℃,氢油体积比500-1200nm3/m3,液时体积空速2.0-4.0h-1
;所述加氢异构化反应的氢分压4-7mpa,反应温度260-370℃,氢油体积比300-500nm3/m3,液时体积空速1.0-2.5h-1
。
18.作为本发明的进一步改进,所述催化裂解反应的温度500-550℃、催化剂与原料油的重量比3-5:1,再生温度650-720℃、绝对压力0.2-0.4mpa、水蒸汽与原料油的重量比0.2-0.5:1。
19.本发明具有如下有益效果:本发明加氢精制过程中,催化裂化柴油中含有大量的多环芳烃、含硫化合物等,pt和pd等贵金属在催化其加氢反应时容易失活,因此,mo和w这种过渡金属作为加氢活性组分,对s元素不敏感,同时,具有很好的催化加氢精制的活性。通过引入少量的b,使催化剂布朗斯特酸量增强,提高加氢脱硫、脱氮活性;本发明加氢精制催化剂催化剂在硫组分存在条件下,稳定性佳,通过b元素的掺杂的mo-w沉积褶皱石墨包覆纳米球催化剂;通过制备一种褶皱石墨包覆氧化硅纳米球的载体结构,褶皱化的氧化石墨可以通过增大比表面积提高对mo和w金属的负载量,同时,增大了与原料油的反应面积,大大提高了精制效率,提高了对原料油的脱硫、脱氮活性,使得制得的成品油中s、n含量明显降低。
20.通过对nay型分子筛进行离子交换和乙酸改性,提高催化剂活性、稳定性、使晶胞常数保持最佳值;改变分子筛沸石表面积和晶胞大小以调节生成油中异构烷烃和烯烃之比;改变分子筛晶粒大小,提高对汽油的选择性同时改变分子筛晶体内表面的可接近性。
21.本发明加氢异构化催化剂为ni-v的磷化物沉积改性hy型分子筛催化剂;在合适的ni、v负载量时,可以保持很好的焦炭和干气的选择性,担载贵金属的催化剂虽然具有高的异构化选择性,但是催化剂的成本较高,而且长时间使用后金属纳米晶因发生聚集而使催化性能下降。以ni等非贵金属为金属位的催化剂尽管成本较低,但是加氢异构化反应的选择性低。因此,本发明制备了过渡金属磷化物ni-v的磷化物作为催化活性组分,具有较高的催化活性及良好的化学稳定性,耐受性好、催化活性高,且成本低。
22.本发明催化裂解催化剂为pt-ni金属沉积改性hy型分子筛催化剂活性组分与氧化铝基质材料的混合物制得的催化剂。选择pt、ni作为双金属催化剂活性金属,因为,进行到这一步时,油中几乎不含有s元素,对pt这种贵金属无明显的失活影响,同时,pt金属的引入明显提高了加氢活性,使用ni作为辅助过渡金属,降低了催化剂的制备成本,同时,也能对pt金属的催化具有协同增效的作用。本发明将催化剂活性组分与氧化铝基质材料混合,通过使用氧化铝为基质材料,可以一定程度提高制得的清洁汽油的辛烷值,提高燃烧性能。制得的该催化剂提高了催化剂一次转化能力,在较温和的条件下,有足够的催化裂解效率,同时,能够控制焦炭产率,对汽油辛烷值进行补偿。
23.本发明通过提供的催化裂化柴油加氢生产清洁汽油的方法,采用加氢精制、异构化、裂解反应,以催化裂化柴油为原料,生产高芳烃含量的优质清洁汽油,促进催化裂化柴油完全转化,产品质量优良,产品收率高,汽油馏分具有低的硫含量和烯烃含量,其中芳烃
含量高,主要为单环芳烃,脱除了大部分多环芳烃,作为高辛烷值汽油产品,同时制备过程中,整体能耗低,降低了设备投资,显著提升催化裂化柴油价值,具有广泛的应用前景。
具体实施方式
24.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
25.氧化石墨为市售或自行制备,制备方法如下:步骤一、称取天然石墨粉10g,过硫酸钾4g,五氧化二磷10g,加入到装有25ml硫酸的反应釜中,60℃恒温水浴中搅拌反应3h,然后移入25℃的恒温水浴中搅拌反应5h,抽滤,水清洗到中性,干燥,得到预氧化石墨;步骤二、称取lg的预氧化石墨,加入到装有25ml硫酸的反应釜中,放入冰水浴中,全部溶解以后,加入3g的高锰酸钾,搅拌反应2h,再移至35℃的恒温水浴中搅拌反应40min,最后加入去离子水,继续35搅拌反应1h,最后滴加30wt%的h2o2,至不再有气体生成为止,溶液变为亮黄色。趁热离心过滤,用5wt%盐酸和去离子水清洗至中性。将终沉淀物超声震荡1h后,干燥24h,得到氧化石墨。
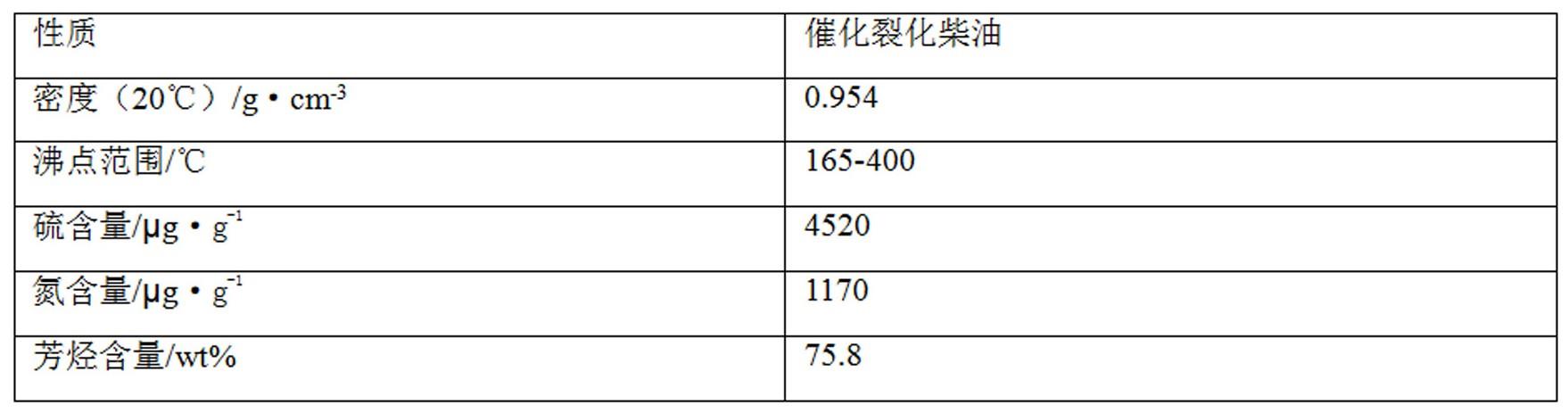
26.催化裂化柴油的性质如表1。
27.表1。
28.制备例1 加氢精制催化剂的制备方法如下:s1.sio2纳米球的制备:将10重量份正硅酸甲酯溶于100重量份石油醚中,加入200重量份含有1重量份十二烷基苯磺酸钠的水溶液中,调节溶液ph值为9,12000r/min乳化10min,加热至60℃,搅拌反应3h,离心,洗涤,干燥,制得sio2纳米球;s2.褶皱氧化石墨包覆纳米球的制备:将12重量份氧化石墨溶于100重量份水中,加入10重量份步骤s1制得的sio2纳米球,搅拌混合10min,喷雾干燥,制得褶皱氧化石墨包覆纳米球;所述喷雾干燥的条件为进风温度为90℃,出风温度为30℃,蒸发水量为1700ml/h;s3.mo-w金属的沉积:将1重量份5.5mol/l浓硝酸加入100重量份含10重量份硝酸钼溶液中,得到a液;将12重量份钨酸铵溶于100重量份水中,得到b液;将111重量份a液加入112重量份b液中,加入25重量份步骤s2制得的褶皱氧化石墨包覆纳米球,搅拌进行共沉淀反应0.5h,过滤,250℃焙烧2h,制得mo-w氧化物沉积褶皱氧化石墨包覆纳米球;s4.还原:将步骤s3制得的mo-w氧化物沉积褶皱氧化石墨包覆纳米球,在650℃通
入氢气还原2h,氢气通气量为15l/min,得到mo-w沉积褶皱石墨包覆纳米球;s5.b元素的掺杂:将10重量份步骤s4制得的mo-w沉积褶皱石墨包覆纳米球加入12重量份3wt%的硼酸钠溶液中,挥干溶剂,干燥,制得加氢精制催化剂。
29.制备例2 加氢精制催化剂的制备方法如下:s1.sio2纳米球的制备:将12重量份正硅酸乙酯溶于100重量份乙酸乙酯中,加入200重量份含有2重量份十四烷基苯磺酸钠的水溶液中,调节溶液ph值为10,15000r/min乳化15min,加热至70℃,搅拌反应5h,离心,洗涤,干燥,制得sio2纳米球;s2.褶皱氧化石墨包覆纳米球的制备:将15重量份氧化石墨溶于100重量份水中,加入10重量份步骤s1制得的sio2纳米球,搅拌混合10min,喷雾干燥,制得褶皱氧化石墨包覆纳米球;所述喷雾干燥的条件为进风温度为100℃,出风温度为50℃,蒸发水量为2200ml/h;s3.mo-w金属的沉积:将2重量份6mol/l浓硝酸加入100重量份含12重量份硝酸钼溶液中,得到a液;将15重量份钨酸铵溶于100重量份水中,得到b液;将114重量份a液加入115重量份b液中,加入30重量份步骤s2制得的褶皱氧化石墨包覆纳米球,搅拌进行共沉淀反应1h,过滤,350℃焙烧3h,制得mo-w氧化物沉积褶皱氧化石墨包覆纳米球;s4.还原:将步骤s3制得的mo-w氧化物沉积褶皱氧化石墨包覆纳米球,在750℃通入氢气还原4h,氢气通气量为20l/min,得到mo-w沉积褶皱石墨包覆纳米球;s5.b元素的掺杂:将10重量份步骤s4制得的mo-w沉积褶皱石墨包覆纳米球加入15重量份5wt%的硼酸铵溶液中,挥干溶剂,干燥,制得加氢精制催化剂。
30.制备例3 加氢精制催化剂的制备方法如下:s1.sio2纳米球的制备:将11重量份正硅酸乙酯溶于100重量份乙酸丁酯中,加入200重量份含有1.5重量份十六烷基磺酸钠的水溶液中,调节溶液ph值为9.5,13500r/min乳化12min,加热至65℃,搅拌反应4h,离心,洗涤,干燥,制得sio2纳米球;s2.褶皱氧化石墨包覆纳米球的制备:将13.5重量份氧化石墨溶于100重量份水中,加入10重量份步骤s1制得的sio2纳米球,搅拌混合10min,喷雾干燥,制得褶皱氧化石墨包覆纳米球;所述喷雾干燥的条件为进风温度为95℃,出风温度为40℃,蒸发水量为2000ml/h;s3.mo-w金属的沉积:将1.5重量份5.7mol/l浓硝酸加入100重量份含11重量份硝酸钼溶液中,得到a液;将13.5重量份钨酸铵溶于100重量份水中,得到b液;将112.5重量份a液加入113.5重量份b液中,加入27重量份步骤s2制得的褶皱氧化石墨包覆纳米球,搅拌进行共沉淀反应1h,过滤,300℃焙烧2.5h,制得mo-w氧化物沉积褶皱氧化石墨包覆纳米球;s4.还原:将步骤s3制得的mo-w氧化物沉积褶皱氧化石墨包覆纳米球,在700℃通入氢气还原3h,氢气通气量为17l/min,得到mo-w沉积褶皱石墨包覆纳米球;s5.b元素的掺杂:将10重量份步骤s4制得的mo-w沉积褶皱石墨包覆纳米球加入13.5重量份4wt%的硼酸钾溶液中,挥干溶剂,干燥,制得加氢精制催化剂。
31.对比制备例1
与制备例3相比,不同之处在于,未进行步骤s2。
32.具体如下:s1.sio2纳米球的制备:将11重量份正硅酸乙酯溶于100重量份乙酸丁酯中,加入200重量份含有1.5重量份十六烷基磺酸钠的水溶液中,调节溶液ph值为9.5,13500r/min乳化12min,加热至65℃,搅拌反应4h,离心,洗涤,干燥,制得sio2纳米球;s2.mo-w金属的沉积:将1.5重量份5.7mol/l浓硝酸加入100重量份含11重量份硝酸钼溶液中,得到a液;将13.5重量份钨酸铵溶于100重量份水中,得到b液;将112.5重量份a液加入113.5重量份b液中,加入27重量份步骤1制得的sio2纳米球,搅拌进行共沉淀反应1h,过滤,300℃焙烧2.5h,制得mo-w氧化物沉积sio2纳米球;s3.还原:将步骤s2制得的mo-w氧化物沉积sio2纳米球,在700℃通入氢气还原3h,氢气通气量为17l/min,得到mo-w沉积sio2纳米球;s4.b元素的掺杂:将10重量份步骤s3制得的mo-w沉积sio2纳米球加入13.5重量份4wt%的硼酸钾溶液中,挥干溶剂,干燥,制得加氢精制催化剂。
33.对比制备例2与制备例3相比,不同之处在于,步骤s3中未添加钨酸铵。
34.具体如下:s3.mo金属的沉积:将1.5重量份5.7mol/l浓硝酸加入100重量份含24.5重量份硝酸钼溶液中加入27重量份步骤s2制得的褶皱氧化石墨包覆纳米球,挥干溶剂,过滤,300℃焙烧2.5h,制得mo氧化物沉积褶皱氧化石墨包覆纳米球。
35.对比制备例3与制备例3相比,不同之处在于,步骤s3中未添加硝酸钼。
36.具体如下:s3.w金属的沉积:将24.5重量份钨酸铵溶于100重量份水中,加入27重量份步骤s2制得的褶皱氧化石墨包覆纳米球,挥干溶剂,过滤,300℃焙烧2.5h,制得w氧化物沉积褶皱氧化石墨包覆纳米球。
37.对比制备例4与制备例3相比,不同之处在于,未进行步骤s3。
38.具体如下:s1.sio2纳米球的制备:将11重量份正硅酸乙酯溶于100重量份乙酸丁酯中,加入200重量份含有1.5重量份十六烷基磺酸钠的水溶液中,调节溶液ph值为9.5,13500r/min乳化12min,加热至65℃,搅拌反应4h,离心,洗涤,干燥,制得sio2纳米球;s2.褶皱氧化石墨包覆纳米球的制备:将13.5重量份氧化石墨溶于100重量份水中,加入10重量份步骤s1制得的sio2纳米球,搅拌混合10min,喷雾干燥,制得褶皱氧化石墨包覆纳米球;所述喷雾干燥的条件为进风温度为95℃,出风温度为40℃,蒸发水量为2000ml/h;s3.还原:将步骤s2制得的褶皱氧化石墨包覆纳米球,在700℃通入氢气还原3h,氢气通气量为17l/min,得到褶皱石墨包覆纳米球;s4.b元素的掺杂:将10重量份步骤s3制得的褶皱石墨包覆纳米球加入13.5重量份4wt%的硼酸钾溶液中,挥干溶剂,干燥,制得加氢精制催化剂。
39.对比制备例5与制备例3相比,不同之处在于,未进行步骤s5。
40.具体如下:s1.sio2纳米球的制备:将11重量份正硅酸乙酯溶于100重量份乙酸丁酯中,加入200重量份含有1.5重量份十六烷基磺酸钠的水溶液中,调节溶液ph值为9.5,13500r/min乳化12min,加热至65℃,搅拌反应4h,离心,洗涤,干燥,制得sio2纳米球;s2.褶皱氧化石墨包覆纳米球的制备:将13.5重量份氧化石墨溶于100重量份水中,加入10重量份步骤s1制得的sio2纳米球,搅拌混合10min,喷雾干燥,制得褶皱氧化石墨包覆纳米球;所述喷雾干燥的条件为进风温度为95℃,出风温度为40℃,蒸发水量为2000ml/h;s3.mo-w金属的沉积:将1.5重量份5.7mol/l浓硝酸加入100重量份含11重量份硝酸钼溶液中,得到a液;将13.5重量份钨酸铵溶于100重量份水中,得到b液;将112.5重量份a液加入113.5重量份b液中,加入27重量份步骤s2制得的褶皱氧化石墨包覆纳米球,搅拌进行共沉淀反应1h,过滤,300℃焙烧2.5h,制得mo-w氧化物沉积褶皱氧化石墨包覆纳米球;s4.还原:将步骤s3制得的mo-w氧化物沉积褶皱氧化石墨包覆纳米球,在700℃通入氢气还原3h,氢气通气量为17l/min,得到mo-w沉积褶皱石墨包覆纳米球,即为加氢精制催化剂。
41.测试例1采用3-flex 3500多站高通量气体吸附仪测定制备例1-3和对比例1-5制得的加氢精制催化剂的比表面积,结果见表2。
42.表2。
43.由上表可知,本发明制备例1-3制得的加氢精制催化剂比表面积大,总孔容大。
44.制备例4 加氢异构化催化剂的制备方法如下:t1.改性hy型分子筛的制备:将10重量份nay型分子筛加入100重量份7wt%的硫酸铵溶液中,搅拌进行离子交换反应1h,过滤,洗涤,加入100重量份2mol/l的乙酸溶液中,加热至70℃,搅拌反应2h,洗涤,500℃焙烧1h,得到改性hy型分子筛;t2.p-ni-v混合前驱溶液的制备:将5重量份硝酸镍、3重量份二氯氧钒溶于100重量份水中,加入10重量份磷酸氢二铵,搅拌混合20min,得到p-ni-v混合前驱溶液;t3.加氢异构化催化剂的制备:将10重量份步骤t1制得的改性hy型分子筛加入30
重量份步骤t2制得的p-ni-v混合前驱溶液中,挥干溶剂,以10℃/min升到250℃,再以1℃/min升到650℃后保持2h进行氢气还原,氢气通气量为15l/min,制得加氢异构化催化剂。
45.制备例5 加氢异构化催化剂的制备方法如下:t1.改性hy型分子筛的制备:将10重量份nay型分子筛加入100重量份10wt%的硫酸铵溶液中,搅拌进行离子交换反应3h,过滤,洗涤,加入100重量份4mol/l的乙酸溶液中,加热至80℃,搅拌反应4h,洗涤,600℃焙烧3h,得到改性hy型分子筛;t2.p-ni-v混合前驱溶液的制备:将7重量份硝酸镍、5重量份二氯氧钒溶于100重量份水中,加入12重量份磷酸氢二铵,搅拌混合20min,得到p-ni-v混合前驱溶液;t3.加氢异构化催化剂的制备:将12重量份步骤t1制得的改性hy型分子筛加入50重量份步骤t2制得的p-ni-v混合前驱溶液中,挥干溶剂,以12℃/min升到250℃,再以1℃/min升到650℃后保持3h进行氢气还原,氢气通气量为15l/min,制得加氢异构化催化剂。
46.制备例6 加氢异构化催化剂的制备方法如下:t1.改性hy型分子筛的制备:将10重量份nay型分子筛加入100重量份8.5wt%的硫酸铵溶液中,搅拌进行离子交换反应2h,过滤,洗涤,加入100重量份3mol/l的乙酸溶液中,加热至75℃,搅拌反应3h,洗涤,550℃焙烧2h,得到改性hy型分子筛;t2.p-ni-v混合前驱溶液的制备:将6重量份硝酸镍、4重量份二氯氧钒溶于100重量份水中,加入11重量份磷酸氢二铵,搅拌混合20min,得到p-ni-v混合前驱溶液;t3.加氢异构化催化剂的制备:将11重量份步骤t1制得的改性hy型分子筛加入40重量份步骤t2制得的p-ni-v混合前驱溶液中,挥干溶剂,以11℃/min升到250℃,再以1℃/min升到650℃后保持2.5h进行氢气还原,氢气通气量为15l/min,制得加氢异构化催化剂。
47.对比制备例6与制备例6相比,不同之处在于,未进行步骤t1。
48.具体如下:t1.p-ni-v混合前驱溶液的制备:将6重量份硝酸镍、4重量份二氯氧钒溶于100重量份水中,加入11重量份磷酸氢二铵,搅拌混合20min,得到p-ni-v混合前驱溶液;t2.加氢异构化催化剂的制备:将11重量份nay型分子筛加入40重量份步骤t2制得的p-ni-v混合前驱溶液中,挥干溶剂,以11℃/min升到250℃,再以1℃/min升到650℃后保持2.5h进行氢气还原,氢气通气量为15l/min,制得加氢异构化催化剂。
49.对比制备例7与制备例6相比,不同之处在于,步骤t2中未添加硝酸镍。
50.具体如下:t2.p-v混合前驱溶液的制备:将10重量份二氯氧钒溶于100重量份水中,加入11重量份磷酸氢二铵,搅拌混合20min,得到p-v混合前驱溶液。
51.对比制备例8与制备例6相比,不同之处在于,步骤t2中未添加二氯氧钒。
52.具体如下:t2.p-ni混合前驱溶液的制备:将10重量份硝酸镍溶于100重量份水中,加入11重
量份磷酸氢二铵,搅拌混合20min,得到p-ni混合前驱溶液。
53.对比制备例9与制备例6相比,不同之处在于,步骤t2中未添加硝酸镍和二氯氧钒。
54.具体如下:t2.p前驱溶液的制备:将21重量份磷酸氢二铵溶于100重量份水中,搅拌混合20min,得到p前驱溶液。
55.对比制备例10与制备例6相比,不同之处在于,步骤t2中未添加磷酸氢二铵。
56.具体如下:t2.ni-v混合前驱溶液的制备:将6重量份硝酸镍、4重量份二氯氧钒溶于100重量份水中,搅拌混合20min,得到ni-v混合前驱溶液。
57.测试例2采用3-flex 3500多站高通量气体吸附仪测定制备例4-6和对比例6-10制得的加氢异构化催化剂的比表面积、孔容及孔结构,结果见表3。
58.表3。
59.由上表可知,本发明制备例4-6制得的加氢异构化催化剂比表面积大,总孔容大。
60.制备例7 催化裂解催化剂的制备方法如下:u1.改性hy型分子筛的制备:将10重量份nay型分子筛加入100重量份7wt%的硫酸铵溶液中,搅拌进行离子交换反应1h,过滤,洗涤,加入100重量份2mol/l的乙酸溶液中,加热至70℃,搅拌反应2h,洗涤,500℃焙烧1h,得到改性hy型分子筛;u2.pt-ni金属沉积:将5重量份硝酸镍、3重量份氯化铂溶于100重量份水中,加入25重量份步骤u1制得的改性hy型分子筛,挥干溶剂,以5℃/min的升温速率升温至400℃下恒温氢气还原2h,氢气通气量为15l/min,制得催化剂活性成分;u3.催化裂解催化剂的制备:将拟薄水铝石在450℃焙烧1h,粉碎,制得基质材料,将50重量份基质材料和5重量份催化剂活性成分混合均匀,制得催化裂解催化剂。
61.制备例8 催化裂解催化剂的制备方法如下:u1.改性hy型分子筛的制备:将10重量份nay型分子筛加入100重量份7wt%的硫酸铵溶液中,搅拌进行离子交换反应1h,过滤,洗涤,加入100重量份2mol/l的乙酸溶液中,加
热至70℃,搅拌反应2h,洗涤,500℃焙烧1h,得到改性hy型分子筛;u2.pt-ni金属沉积:将7重量份硝酸镍、5重量份氯化铂溶于100重量份水中,加入30重量份步骤u1制得的改性hy型分子筛,挥干溶剂,以7℃/min的升温速率升温至500℃下恒温氢气还原4h,氢气通气量为15l/min,制得催化剂活性成分;u3.催化裂解催化剂的制备:将拟薄水铝石在550℃焙烧3h,粉碎,制得基质材料,将60重量份基质材料和7重量份催化剂活性成分混合均匀,制得催化裂解催化剂。
62.制备例9 催化裂解催化剂的制备方法如下:u1.改性hy型分子筛的制备:将10重量份nay型分子筛加入100重量份8.5wt%的硫酸铵溶液中,搅拌进行离子交换反应2h,过滤,洗涤,加入100重量份3mol/l的乙酸溶液中,加热至75℃,搅拌反应3h,洗涤,550℃焙烧2h,得到改性hy型分子筛;u2.pt-ni金属沉积:将6重量份硝酸镍、4重量份氯化铂溶于100重量份水中,加入27重量份步骤u1制得的改性hy型分子筛,挥干溶剂,以6℃/min的升温速率升温至450℃下恒温氢气还原3h,氢气通气量为15l/min,制得催化剂活性成分;u3.催化裂解催化剂的制备:将拟薄水铝石在500℃焙烧2h,粉碎,制得基质材料,将55重量份基质材料和6重量份催化剂活性成分混合均匀,制得催化裂解催化剂。
63.对比制备例11与制备例9相比,不同之处在于,未进行步骤u1。
64.具体如下:u1.pt-ni金属沉积:将6重量份硝酸镍、4重量份氯化铂溶于100重量份水中,加入27重量份nay型分子筛,挥干溶剂,以6℃/min的升温速率升温至450℃下恒温氢气还原3h,氢气通气量为15l/min,制得催化剂活性成分;u2.催化裂解催化剂的制备:将拟薄水铝石在500℃焙烧2h,粉碎,制得基质材料,将55重量份基质材料和6重量份催化剂活性成分混合均匀,制得催化裂解催化剂。
65.对比制备例12与制备例9相比,不同之处在于,步骤u2中未添加硝酸镍。
66.具体如下:u2.pt金属沉积:将10重量份氯化铂溶于100重量份水中,加入27重量份步骤u1制得的改性hy型分子筛,挥干溶剂,以6℃/min的升温速率升温至450℃下恒温氢气还原3h,氢气通气量为15l/min,制得催化剂活性成分。
67.对比制备例13与制备例9相比,不同之处在于,步骤u2中未添加氯化铂。
68.具体如下:u2.ni金属沉积:将10重量份硝酸镍氯化铂溶于100重量份水中,加入27重量份步骤u1制得的改性hy型分子筛,挥干溶剂,以6℃/min的升温速率升温至450℃下恒温氢气还原3h,氢气通气量为15l/min,制得催化剂活性成分。
69.对比制备例14与制备例9相比,不同之处在于,步骤u2中未添加硝酸镍和氯化铂。
70.具体如下:
u2.pt-ni金属沉积:将27重量份步骤u1制得的改性hy型分子筛,挥干溶剂,以6℃/min的升温速率升温至450℃下恒温氢气还原3h,氢气通气量为15l/min,制得催化剂活性成分。
71.对比制备例15与制备例9相比,不同之处在于,步骤u3中基质材料由等量的高岭土替代。
72.具体如下:u3.催化裂解催化剂的制备:将55重量份高岭土和6重量份催化剂活性成分混合均匀,制得催化裂解催化剂。
73.实施例1:
74.本实施例提供一种催化裂化柴油加氢生产清洁汽油的方法,包括以下步骤:(1)将催化裂化柴油(催化裂化柴油原料的沸点范围为165-400℃,总芳烃含量高于60wt%)与氢气混合,在制备例1制得的加氢精制催化剂的催化作用下,进行加氢精制反应;所述加氢精制反应的氢分压5-12mpa,反应温度240-400℃,氢油体积比500-1200nm3/m3,液时体积空速2.0-4.0h-1
;(2)将步骤(1)获得的生成油与氢气混合,在制备例4制得的加氢异构化催化剂的催化作用下,进行加氢异构化反应;所述加氢异构化反应的氢分压4-7mpa,反应温度260-370℃,氢油体积比300-500nm3/m3,液时体积空速1.0-2.5h-1
;(3)将步骤(2)获得的生成油在制备例7制得的催化裂解催化剂作用下发生催化裂解反应,生成低碳烯烃和汽油馏分,产物中》200℃的馏分重新返回步骤(2)进行催化转化;所述催化裂解反应的温度500-550℃、催化剂与原料油的重量比3-5:1,再生温度650-720℃、绝对压力0.2-0.4mpa、水蒸汽与原料油的重量比0.2-0.5:1;(4)收集步骤(3)中的汽油馏分混合均匀后,检验,得到合格清洁汽油产品。
75.实施例2:
76.本实施例提供一种催化裂化柴油加氢生产清洁汽油的方法,包括以下步骤:(1)将催化裂化柴油(催化裂化柴油原料的沸点范围为165-400℃,总芳烃含量高于60wt%)与氢气混合,在制备例2制得的加氢精制催化剂的催化作用下,进行加氢精制反应;所述加氢精制反应的氢分压5mpa,反应温度240℃,氢油体积比500nm3/m3,液时体积空速2.0h-1
;(2)将步骤(1)获得的生成油与氢气混合,在制备例5制得的加氢异构化催化剂的催化作用下,进行加氢异构化反应;所述加氢异构化反应的氢分压4mpa,反应温度260℃,氢油体积比300nm3/m3,液时体积空速1.0h-1
;(3)将步骤(2)获得的生成油在制备例8制得的催化裂解催化剂作用下发生催化裂解反应,生成低碳烯烃和汽油馏分,产物中》200℃的馏分重新返回步骤(2)进行催化转化;所述催化裂解反应的温度500℃、催化剂与原料油的重量比3:1,再生温度650℃、绝对压力0.2mpa、水蒸汽与原料油的重量比0.2:1;
(4)收集步骤(3)中的汽油馏分混合均匀后,检验,得到合格清洁汽油产品。
77.实施例3:
78.本实施例提供一种催化裂化柴油加氢生产清洁汽油的方法,包括以下步骤:(1)将催化裂化柴油(催化裂化柴油原料的沸点范围为165-400℃,总芳烃含量高于60wt%)与氢气混合,在制备例3制得的加氢精制催化剂的催化作用下,进行加氢精制反应;所述加氢精制反应的氢分压10mpa,反应温度340℃,氢油体积比800nm3/m3,液时体积空速3.0h-1
;(2)将步骤(1)获得的生成油与氢气混合,在制备例6制得的加氢异构化催化剂的催化作用下,进行加氢异构化反应;所述加氢异构化反应的氢分压5.5mpa,反应温度320℃,氢油体积比400nm3/m3,液时体积空速1.9h-1
;(3)将步骤(2)获得的生成油在制备例9制得的催化裂解催化剂作用下发生催化裂解反应,生成低碳烯烃和汽油馏分,产物中》200℃的馏分重新返回步骤(2)进行催化转化;所述催化裂解反应的温度520℃、催化剂与原料油的重量比4:1,再生温度690℃、绝对压力0.3mpa、水蒸汽与原料油的重量比0.35:1;(4)收集步骤(3)中的汽油馏分混合均匀后,检验,得到合格清洁汽油产品。
79.对比例1-15与实施例3相比,不同之处在于,相应的催化剂分别由对比制备例1-15制得。
80.测试例1将本发明实施例1-3和对比例1-15制得的清洁汽油产品进行性能测试,结果见表4。
81.表4
。
82.由上表可知,本发明实施例1-3制得的清洁汽油产品综合性能更佳。
83.对比制备例1与制备例3相比,未进行步骤s2。硫含量提高,抗爆指数下降,辛烷值下降。本发明褶皱化的氧化石墨可以通过增大比表面积提高对mo和w金属的负载量,同时,增大了与原料油的反应面积,大大提高了精制效率。
84.对比制备例2、3与制备例3相比,步骤s3中未添加钨酸铵或硝酸钼。对比制备例4与制备例3相比,未进行步骤s3。烯烃含量下降,抗爆指数下降,辛烷值下降。mo和w这种过渡金属作为加氢活性组分,对s元素不敏感,同时,具有很好的催化加氢精制的活性,通过制备一种褶皱石墨包覆氧化硅纳米球的载体结构,增大了与原料油的反应面积,大大提高了精制效率,提高了对原料油的脱硫、脱氮活性,使得制得的成品油中s、n含量明显降低。
85.对比制备例5与制备例3相比,未进行步骤s5。硫含量提高,抗爆指数下降。本发明通过引入少量的b,使催化剂布朗斯特酸量增强,提高加氢脱硫、脱氮活性。
86.对比制备例6与制备例6相比,未进行步骤t1。烯烃含量下降,抗爆指数下降,辛烷值下降。通过对nay型分子筛进行离子交换和乙酸改性,提高催化剂活性、稳定性、使晶胞常数保持最佳值;改变分子筛沸石表面积和晶胞大小以调节生成油中异构烷烃和烯烃之比;改变分子筛晶粒大小,提高对汽油的选择性同时改变分子筛晶体内表面的可接近性。
87.对比制备例7、8与制备例6相比,步骤t2中未添加硝酸镍或二氯氧钒。对比制备例9与制备例6相比,步骤t2中未添加硝酸镍和二氯氧钒。烯烃含量下降,抗爆指数下降,辛烷值下降。本发明加氢异构化催化剂为ni-v的磷化物沉积改性hy型分子筛催化剂;在合适的ni、v负载量时,可以保持很好的焦炭和干气的选择性,担载贵金属的催化剂虽然具有高的异构
化选择性,但是催化剂的成本较高,而且长时间使用后金属纳米晶因发生聚集而使催化性能下降。
88.对比制备例10与制备例6相比,步骤t2中未添加磷酸氢二铵。烯烃含量下降,抗爆指数下降,辛烷值下降。以ni等非贵金属为金属位的催化剂尽管成本较低,但是加氢异构化反应的选择性低。因此,本发明制备了过渡金属磷化物ni-v的磷化物作为催化活性组分,具有较高的催化活性及良好的化学稳定性,耐受性好、催化活性高,且成本低。
89.对比制备例11与制备例6相比,未进行步骤u1。烯烃含量下降,芳烃含量下降,抗爆指数下降,辛烷值下降。通过对nay型分子筛进行离子交换和乙酸改性,提高催化剂活性、稳定性、使晶胞常数保持最佳值;改变分子筛沸石表面积和晶胞大小以调节生成油中异构烷烃和烯烃之比;改变分子筛晶粒大小,提高对汽油的选择性同时改变分子筛晶体内表面的可接近性。
90.对比制备例12、13与制备例6相比,步骤u2中未添加硝酸镍或氯化铂。对比制备例14与制备例6相比,步骤u2中未添加硝酸镍和氯化铂。烯烃含量下降,芳烃含量下降,抗爆指数下降,辛烷值下降。本发明催化裂解催化剂为pt-ni金属沉积改性hy型分子筛催化剂活性组分与氧化铝基质材料的混合物制得的催化剂。选择pt、ni作为双金属催化剂活性金属,因为,进行到这一步时,油中几乎不含有s元素,对pt这种贵金属无明显的失活影响,同时,pt金属的引入明显提高了加氢活性,使用ni作为辅助过渡金属,降低了催化剂的制备成本,同时,也能对pt金属的催化具有协同增效的作用。
91.对比制备例15与制备例6相比,步骤u3中基质材料由等量的高岭土替代。抗爆指数下降,辛烷值下降。本发明将催化剂活性组分与氧化铝基质材料混合,通过使用氧化铝为基质材料,可以一定程度提高制得的清洁汽油的辛烷值,提高燃烧性能。制得的该催化剂提高了催化剂一次转化能力,在较温和的条件下,有足够的催化裂解效率,同时,能够控制焦炭产率,对汽油辛烷值进行补偿。
92.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
当前第1页1
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!